單原子催化劑因其最大的原子使用率和電子狀態的高度可調性,成爲能源材料研究領域的熱點,例如過渡金屬單原子催化劑在電催化CO2還原上表現出優異特性,同時,單原子催化劑也被認爲是在原子水平上揭示催化機制的理想模型系統。然而,單原子催化劑的實際應用受到穩定性差和轉換效率低的限制,比如,用于CO2還原(CO2RR)的氮摻雜石墨烯上的鎳單原子爲CO生成提供了高催化活性和法拉第效率,但由于形成 *COOH 中間體的高能量勢壘,其開啓電位較大;鐵單原子對CO2還原的開啓電位較低,但鐵位點與反應中間體(如*CO)的強結合嚴重降低了法拉第效率;過渡金屬位點與給電子中間體的結合強度太強,同樣會降低析氧反應的催化活性等等。
與單原子相比,異類雙原子位點催化劑是利用兩個相鄰的不同金屬原子來實現它們的功能互補和協同作用,特別是,與中間體的結合能可以通過兩個相鄰的異類金屬之間的電子相互作用來調整,克服了單原子催化的不足。然而,合理地構建具有高催化性能和多功能性的雙原子位點催化劑並揭示其工作機制仍然是一個巨大的挑戰。
近日,中山大學材料科學與工程學院曾志平副教授研究組構建了在氮摻雜石墨烯上的高密度鎳鐵雙原子高性能電催化體系,並對催化增強機制進行了深入的研究。他們通過光電子光譜,包括XAS、UPS和XPS,鑒定了兩個異類金屬原子的電子結構特征。X射線吸收近邊結構和擴展X射線吸收精細結構測量證實了鎳鐵雙單原子的鎳電子態具有與單原子相似鎳氮配位鍵,但其中鐵電子態不同于鐵單原子,呈現更高的K-edge電子結構,即更高的氧化態;小波變換擴展X射線吸收圖譜顯示了金屬-氮和金屬-金屬鍵的存在,進一步證實了獨特的雙原子結構。理論計算進一步探究和證實了雙金屬原子對之間的d-d軌道耦合對二氧化碳還原和析氧的影響。鎳和鐵之間的d軌道(尤其是dz2, dx2-y2, 和dxz)相互作用,導致軌道能級下降和電子分布的離散性,從而有益于*CO中間體的脫附。異類雙原子體系中,配位氮pz軌道在費米能級附近的電子態密度增加,得益于軌道耦合中鐵電子轉移至氮的pz軌道,使得鐵活性中心的價態增加,從而有利于CO2RR和析氧反應。異類雙原子催化劑結合了鐵單原子(較低的開啓電位)和鎳單原子(較高的選擇性催化)兩者的優勢,表現出遠優于對應的單原子催化劑的卓越電催化活性、高度的選擇性和穩定性。配備該催化劑的金屬-CO2電池可在大電流密度下穩定和持續地充放電,在CO2還原的同時獲取電能和燃料(CO,CH4, H2等),具有很好的實用性前景,有望拓展至火星(大氣CO2濃度95%)探索。
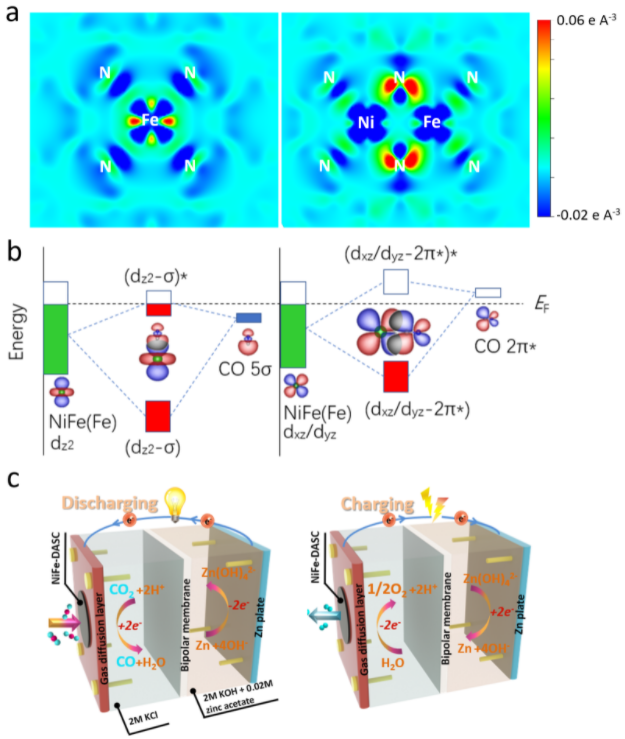
圖1. Fe單原子(Fe-SAC)和FeNi 雙原子位點(FeNi-DASC)催化劑中(a)Fe位點電荷分布,(b)Fe位點3d 軌道于吸附的CO (5σ and 2π* 軌道相互作用示意圖,(c)FeNi-DASC的金屬-CO2電池充放電過程。
該研究成果論文“Orbital coupling of hetero-diatomic nickel-iron site for bifunctional electrocatalysis of CO2 reduction and oxygen evolution”發表在國際重要學術期刊Nature Communications,曾志平副教授是論文的第一作者,新加坡南洋理工大學陳鵬教授、劉彬教授、蘇州科技大學楊鴻斌教授爲文章的共同通訊作者。該研究工作得到了國家自然科學基金委員會、中山大學“百人計劃”啓動經費和新加坡科技局、新加坡教育部的支持。
來源:中山大學
論文鏈接:
https://www.nature.com/articles/s41467-021-24052-5