爲了實現精准醫療,許多國家已經開展本地人群的全基因組測序(WGS)研究。不過,目前的WGS資源主要來自歐洲人,而亞洲基因組在公共數據庫中的代表性不足。
爲此,新加坡科技研究局(A*STAR)領導的團隊對新加坡的中國人、馬來人和印度人三個群體開展了大規模測序,並在《Cell》雜志上發表了題爲“Large-Scale Whole-Genome Sequencing of Three Diverse Asian Populations in Singapore”的文章。
“新加坡三個主要種族提供了東南亞地區遺傳多樣性的獨特快照,”作者在文中寫道。這篇文章的通訊作者包括華中科技大學的王超龍教授和新加坡國立大學的劉建軍教授,他們同時隸屬于新加坡科技研究局。
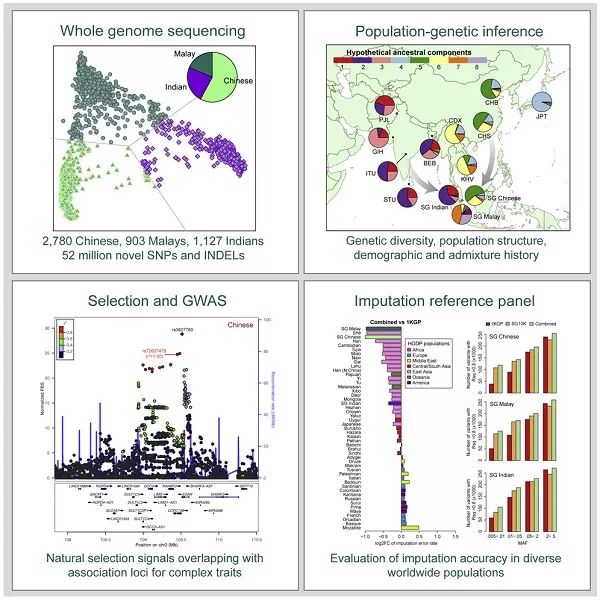
SG10K聯盟的成員對新加坡的4,810名中國人、馬來人和印度人進行了測序,發現了9,830萬個新穎的或已知的多態性,可用來評估亞洲血統在這些族裔中的比例。他們還追蹤了至少20個表現出自然選擇迹象的基因座,其中14個與複雜性狀和疾病相關聯。
基于這些研究,作者認爲“對新加坡人的全基因組測序有望讓整個亞洲以及世界其他地區的人群受益”。他們預計,“SG10K的數據將成爲寶貴的資源,可推動亞洲人的遺傳性狀和複雜疾病研究,並緩解目前人類遺傳學研究中的群體差異”。
這項研究屬于新加坡萬人基因組測序計劃的早期階段。在此,研究人員對4,800多人進行測序,平均測度深度達13.7倍。參與者包括2,780名中國人、903名馬來人和1,127名印度人,來自8個不同的隊列。
在剔除了不符合質量控制標准的變異後,他們獲得了8,910萬個SNP和910萬個小的插入缺失(indel)。其中,大約4,560萬個SNP和630萬個indel未曾出現在以往的群體研究中。
研究人員將1,200多名個體以往的基因型數據與此次分析的結果進行比較,確認了這些變異的質量,然後才深入研究芯片未覆蓋的罕見變異。通過這種方式,他們記錄了2,525名健康個體的變異數量和類型,包括與隱性疾病風險相關的變異。
通過主成分分析(PCA),研究人員發現新加坡中國人和新加坡印度人分別與東亞人和南亞人重疊,而新加坡馬來人與其他群體相對較遠,這意味著馬來人代表了東南亞的土著人。同時,他們還追溯了新加坡各個人群的起源。
考慮到SG10K的數據將成爲亞洲人群數據的寶貴資源,研究人員接著評估了數據集的有效性。“即使采用嚴格的標准,我們也能夠檢測到亞洲人群以往報道的許多基因座,”他們寫道。這些結果凸顯了SG10K數據將成爲推動亞洲人群遺傳學研究的寶貴資源。
原文檢索
Large-Scale Whole-Genome Sequencing of Three Diverse Asian Populations in Singapore
Cell VOLUME 179, ISSUE 3, P736-749.E15, OCTOBER 17, 2019
